


Anti-SDHA: Rabbit Succinate Dehydrogenase A Antibody |
 |
BACKGROUND The succinate dehydrogenase (SDH) (succinate: ubiquinone oxidoreductase) is a mitochondrial enzyme complex (complex II) with an important role in oxydative phosphorylation and intracellular oxygene sensing and signaling. SDH catalyses the oxidation of succinate into fumarate in the Krebs cycle, derived electrons being fed to the respiratory chain complex III to reduce oxygen and form water. This builds up an electrochemical gradient across the mitochondrial inner membrane allowing for the synthesis of ATP. Alternatively, electrons can be diverted to reduce the ubiquinone pool (UQ pool) and provide reducing equivalents necessary to reduce superoxide anions originating either from an exogenous source or from the respiratory chain itself. A complete lack of succinate dehydrogenase activity will hamper electron flow to both respiratory chain complex III and the quinone pool, resulting in a major oxidative stress known to promote tumor formation in human.1
SDH consists of four nuclearly encoded subunits (SDHA, SDHB, SDHC and SDHD) whose structure and genes being mostly conserved through evolution. SDHA (Ch5p15) and SDHB (Ch1p36) encode the two catalytic subunits, the flavoprotein and the iron-sulfur protein respectively; SDHC (Ch1q21) and SDHD (Ch11q23) encode transmembrane proteins that anchor complex II in the inner mitochondrial membrane, and contain a ubiquinone binding site. Both SDHC and SDHD are often referred to as the anchoring subunits, although they are actually required for electron transfer from succinate to the ubiquinone pool as well. The SDHA gene consists of 15 exons, with a second isoform and at least one pseudogene (Ch3q29) present in the genome. SDHB has eight exons and no known pseudogenes, while SDHC covers six exons and has three candidate pseudogenes. SDHD has four exons and six reported intronless pseudogenes.2 Defects of the succinate dehydrogenase are comparatively rare in human. Yet, large differences in clinical presentations have been reported in patients harboring mutations in one of the four SDH subunit-encoding genes. Mutations in the flavoprotein subunit, SDHA were reported to cause a mitochondrial RC defect and resulted in typical mitochondrial encephalopathy, namely Leigh syndrome, but have not as yet been identified as a factor in hereditary cancer. However, mutations in subunits B, C and D have been shown to cause paraganglioma, generally benign, vascularized tumors in the head and the neck, or phaeochromocytomas. The carotid body, the most common tumor site, is a highly vascular small organ localized at the bifurcation of the common carotid artery and is a chemoreceptive organ that senses oxygen levels in the blood. Because of the occurrence of somatic mutations of these three genes in tumors, they should be regarded as tumor-suppressor genes. So far, SDH is only known to catalyze a unique reaction, which requires the participation of its four subunits, and deleterious mutations in any of the SDH genes should invariably result in a decreased SDH activity.3
SDH consists of four nuclearly encoded subunits (SDHA, SDHB, SDHC and SDHD) whose structure and genes being mostly conserved through evolution. SDHA (Ch5p15) and SDHB (Ch1p36) encode the two catalytic subunits, the flavoprotein and the iron-sulfur protein respectively; SDHC (Ch1q21) and SDHD (Ch11q23) encode transmembrane proteins that anchor complex II in the inner mitochondrial membrane, and contain a ubiquinone binding site. Both SDHC and SDHD are often referred to as the anchoring subunits, although they are actually required for electron transfer from succinate to the ubiquinone pool as well. The SDHA gene consists of 15 exons, with a second isoform and at least one pseudogene (Ch3q29) present in the genome. SDHB has eight exons and no known pseudogenes, while SDHC covers six exons and has three candidate pseudogenes. SDHD has four exons and six reported intronless pseudogenes.2 Defects of the succinate dehydrogenase are comparatively rare in human. Yet, large differences in clinical presentations have been reported in patients harboring mutations in one of the four SDH subunit-encoding genes. Mutations in the flavoprotein subunit, SDHA were reported to cause a mitochondrial RC defect and resulted in typical mitochondrial encephalopathy, namely Leigh syndrome, but have not as yet been identified as a factor in hereditary cancer. However, mutations in subunits B, C and D have been shown to cause paraganglioma, generally benign, vascularized tumors in the head and the neck, or phaeochromocytomas. The carotid body, the most common tumor site, is a highly vascular small organ localized at the bifurcation of the common carotid artery and is a chemoreceptive organ that senses oxygen levels in the blood. Because of the occurrence of somatic mutations of these three genes in tumors, they should be regarded as tumor-suppressor genes. So far, SDH is only known to catalyze a unique reaction, which requires the participation of its four subunits, and deleterious mutations in any of the SDH genes should invariably result in a decreased SDH activity.3
REFERENCES
1. Rustin, P. et al: Eur. J. Hum. Genet. 10:289-91, 2002
2. Bayley, J.P. et al: BMC Med. Genet. 6:39, 2005
3. Pawlu, C. et al: Familial Cancer 4:49-54, 2005
2. Bayley, J.P. et al: BMC Med. Genet. 6:39, 2005
3. Pawlu, C. et al: Familial Cancer 4:49-54, 2005
Products are for research use only. They are not intended for human, animal, or diagnostic applications.
Параметры
Cat.No.: | CA1306 |
Antigen: | Short peptide from human SDHA sequence. |
Isotype: | Rabbit IgG |
Species & predicted species cross- reactivity ( ): | Human, Mouse, Rat |
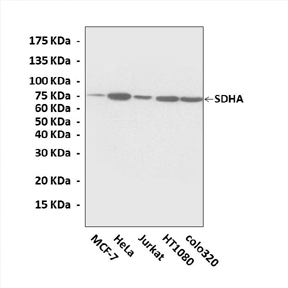
Applications & Suggested starting dilutions:* | WB 1:1000 IP n/d IHC 1:50 - 1:200 ICC n/d FACS n/d |
Predicted Molecular Weight of protein: | 75 kDa |
Specificity/Sensitivity: | Detects endogenous levels of SDHA proteins without cross-reactivity with other related proteins. |
Storage: | Store at -20°C, 4°C for frequent use. Avoid repeated freeze-thaw cycles. |
*Optimal working dilutions must be determined by end user.
Документы

Информация представлена исключительно в ознакомительных целях и ни при каких условиях не является публичной офертой



